
Supramolecular Concepts in Homogeneous Catalysis
Bidentate ligands are important for selectivity control in homogeneous catalysis. However, the quest for the ultimate ligand giving rise to a catalyst with optimal activity and selectivity is difficult. Since rational design still does not allow the ligand of choice for a given reaction and substrate to be predicted, the combinatorial synthesis of ligand libraries and their subsequent use has become an additional strategy. However, the rate-determining step in catalyst development is in most cases the time-consuming ligand synthesis required to generate the library.
As a solution to this problem, and as an alternative to the classical bidentate ligand synthesis, the principle of self-assembly of monodentate to bidentate ligands relying on complementary non-covalent interligand interactions has been introduced by us[50] and others.[66] Thus, inspired by DNA base pairing the method enables rapid generation of catalyst libraries by simple mixing of the components without additional synthetic steps, thus allowing access towards structurally diverse and meaningful ligand libraries (see Scheme 14).
Scheme 14: An A-T base pair model as a complementary platform for specific self-assembly of heterodimeric bidentate ligands.
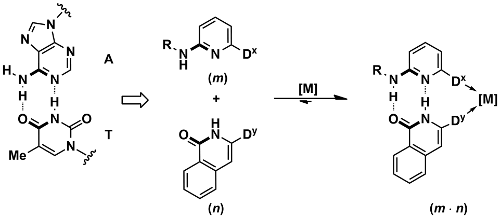
On the basis of this platform, catalyst libraries were generated and screened against a series of transition metal catalyzed reactions. From these studies excellent catalysts for anti-Markovnikov hydration of alkynes,[69] regioselective hydroformylation,[50,61,67,80] the regioselective hydrocyanation of stryrenes,[110] the asymmetric rhodium-catalyzed hydrogenation[70] and the allylation of N-heterocycles with allylic alcohols have emerged.[96] Selected examples are given below (see Scheme 15).
Scheme 15: Examples for the application of supramolcular self-assembly ligand/catalyst libraries.
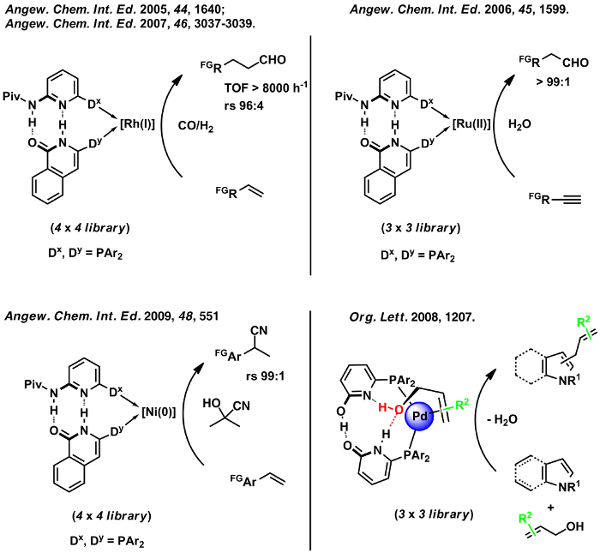
In the case of ruthenium-catalyzed anti-Markovnikov water addition to terminal alkynes [69] as well as for palladium-catalyzed allylation of N-heterocycles employing allylic alcohols as substrates,[96] we collected evidence that the ligand hydrogen-bonding network plays a pivotal role in substrate activation through hydrogen-bonding. This observation led to the development of a new class of phosphine ligands with an additional acyl-guanidinium binding site, which allows for complementary hydrogen-bonding with carboxylic acids (Scheme 16).[93,100,118,122]
Scheme 16: Acylguanidine functionalized phosphine ligands as key components of supramolecular catalysts.
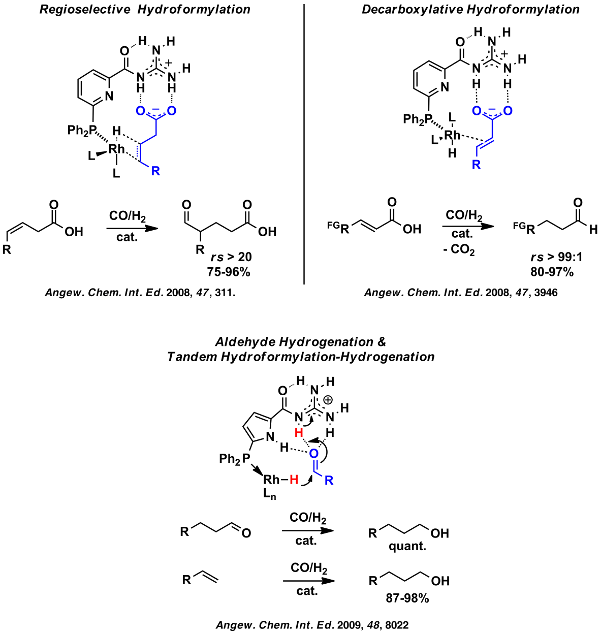
Based on this idea we implemented a new series of catalysts which allow for unusual regioselectivity control upon hydroformylation of terminal and internal α,γ-unsaturated carboxylic acids.[93,122] Furthermore these new catalysts enable the first decarboxylative hydroformylation [100] and showed that they they are capable for highly chemoselective reduction of aldehydes in the course of a Tandem hydroformylation-hydrogenation process (Scheme 16).[118] Studies on the origin of activity and selectivity has revealed that these catalysts indeed rely on additional non-covalent attractive substrate ligand interactions, and thus represent a new generation of catalyst systems with an enzyme-like operation mode. Of particular interest is the mechanism of aldehyde hydrogenation for this research proposal as determined by DFT calculations (Scheme 17).[118]
Scheme 17: Proposed mechanism for aldehyde hydrogenation and corresponding DFT calculations.[118]
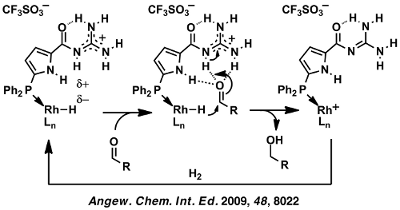
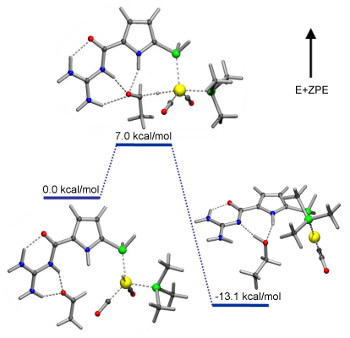
These calculations are in accord with all experimental observations and suggest a reaction mechanism involving carbonyl binding to the acylguanidine function resulting in energy-lowering of the LUMO. A subsequent intramolecular hydride delivery from the rhodium along the Burgi-Dunitz trajectory is the energetically lowest lying pathway for aldehyde reduction. While the transition state involves an additional third pyrrol NH hydrogen-bond, both the starting aldehyde catalyst complex as well as the product alcohol catalyst complex involve two hydrogen bonds. This can be viewed as this catalysts’ specific ability for transition state stabilization. Importantly, it is very reasonable to extrapolate this mode of operation to the activation of other electrophiles and nucleophiles to allow for new carbon/carbon as well as carbon/heteroatom bond forming reactions.